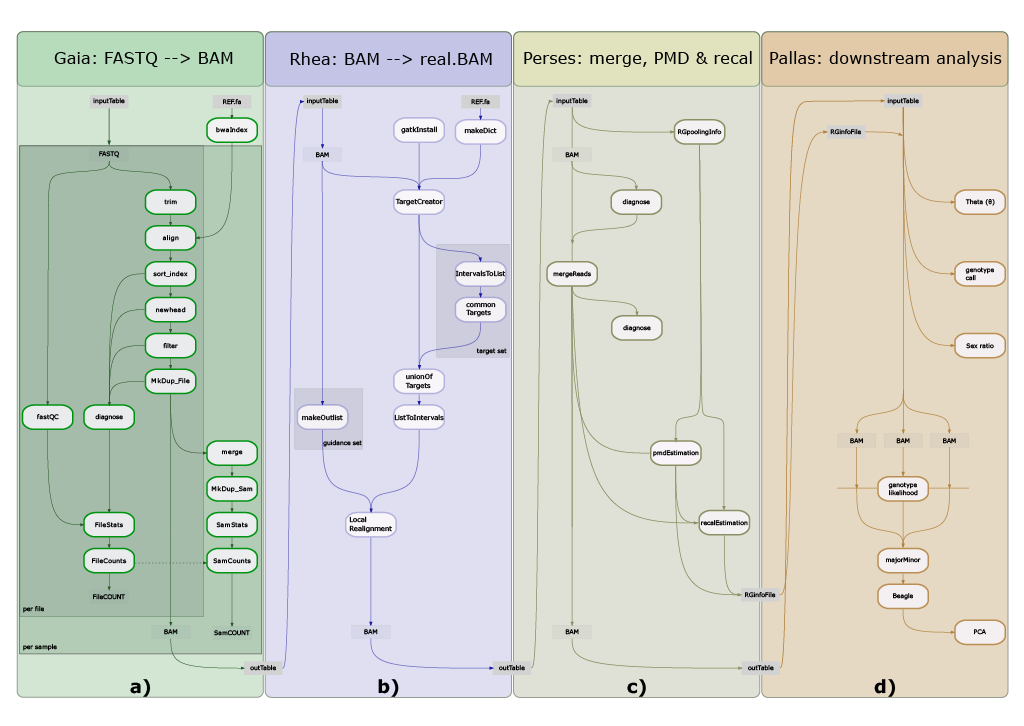
12.5 Pallas
Pallas is the most flexible submodule in the ATLAS-Pipeline.
12.5.1 Input
sample-file
We strongly advise you to run the Perses module, which estimates base quality recalibration parameters and merges paired-end reads before running Pallas.
If you want to use all samples from a previous Perses run, you can define sampleFile: fromPerses. The pipeline will automatically take the produced output-tables as inputfile.
If you want to change the automatic tables to define readgroup-merging, we advise to copy it to another place (like supporting_files/samples_Pallas.tsv) so your changes are not overwritten in case you decide to re-run the Gaia or Rhea pipeline.
If you want to prepare the table by hand (e.g. because you have already filtered BAMfiles or PMD- and recal files at hand), prepare a tab delimited table with the columns as indicated below.
Example: We want to analyze the following files:
/path/to/s1/sample1.bam
/path/to/s2/sample2.bam
/path/to/s3/sample3.bam
/path/to/s4/sample4.bam
/path/to/s5/sample5.bamBecause sample1 and sample2 are of very low depth and did not contain enough data for sufficient PMD and recal analysis, but come from the same sampling site/aera and were sequenced on the same sequencing run, we have estimated the parameters outside of the pipeline for a merged BAMfile from sample1 and sample2. These parameters can now be provided with the optional columns PMDFile and recalFile. In our example, the input file could look similar to this:
| Sample | Path | PMDFile | recalFile |
|---|---|---|---|
| sample1 | /path/to/s1/ | /path/to/PMDfile.txt | /path/to/recalFile.txt |
| sample2 | /path/to/s2/ | /path/to/PMDfile.txt | /path/to/recalFile.txt |
| sample3 | /path/to/s3/ | F | F |
| sample4 | /path/to/s4/ | F | F |
| sample5 | /path/to/s5/ | F | F |
The order of the table columns can be changed. Additional columns can be present. Columns PMDFile and recalFile are optional. The table can contain comments (starting with ‘#’) but no whitespace.
config-file
The config file has to be provided in yaml format. The same example as below can also be found in examples/example_config_Pallas.yaml. To use it as a template, make sure to copy it to a new location, otherwise it will be overwritten once you update the pipeline.
Compulsatory fields are:
- runScript: Pallas
- sampleFile: give location to sample file or specify fromPerses (see also Sample file)
- atlas: location of atlas executable
- ref: location of reference fasta file
runScript: Pallas
sampleFile: fromPerses
atlas: conda
ref: ../Reference/hs37d5.fa
glf: T
maMi: T
###################################################################################################################################
###################################################################################################################################
# optional parameters
# atlasParams: general atlas parameters applying to all jobs unless specified within the job parameters. default: ""
# estimateErrors: if error recalibration from Perses should be considered (T/F). default: "T". Will search for json files in the Perses default location. This location can be updated in the Pallas input file (column estimateErrorsFile)
# parallelize: run glf and major minor tasks in parallel for each chromosome. MajorMinor files will then be concatenated into one outfile. default=F
# binCount: if parallelize=T you can additionally set a number of bins for parallelization. This makes sense if your number of chromosomes/contigs is very high. default=F
# chromPar: if you want to restrict your parallelization on a certain set of chromosomes, add them here as a comma separated string. example: "scaffold_1,scaffold_3,scaffold4". If "F", default=all (all contigs in BAM header considered)
#
# glf: estimate genotype likelihoods. default=F
# glfParams: specific parameters to pass for Atlas-task glf. default: ""
# Attention: do not use option "--chr" if "parallelize" is used! use config parameter "chromPar:" instead.
#
# call: perform base calling with respective method. Can be (Bayes/MLE/AllelePresence/F). default=F
# callParams: specific parameters to pass for Atlas-task call. default: ""
#
# inbreeding: estimate inbreeding coefficient
# inbreedingParams: specific parameters to pass for Atlas-task inbreeding. default: ""
#
# theta: estimate theta either per window or genome wide. Can be (window/genomeWide/F). default=F
# thetaParams: specific parameters to pass for Atlas-task theta. default: ""
#
# maMi: produce major-minor VCF file (will automatically also produce glf-files). default=F
# maMiParams: specific parameters to pass for Atlas-task majorMinor. default: ""
# Attention: do not use option "--chr" if "parallelize" is used! use config parameter "chromPar:" instead.
# Attention: do not use option "--out"! use config parameter "maMiOut:" instead.
# maMiOut: save majorMinor vcf with a different prefix. this can be useful to produce outfiles with different filter options.
# You can also define which maMi file should be used for PCA by providing the prefix (without .vcf.gz) here.
#
# beagle: convert major-minor file to beagle format (as input for ANGSD). default=F
# will automaticaly produce glf-files and major-minor vcf file.
# beagleParams: specific parameters to pass for Atlas-task convertVCF to beagle. default: ""
#
# PCA: perform a PCA with pcANGSD. will automaticaly produce glf-files, major-minor vcf file and beagle file. default=F
# PCAngsd: location of PCAngsd executable. no default. Must be given.
# PCAParams: specific parameters to pass for PCAngsd. default: ""
# threads: threads for PCAngsd. default=8
#
#
###################################################################################
# general parameters
# atlas: specify if Atlas should automatically be loaded from conda whenever necessary (default: conda). If you would like to use a pre-compiled Atlas instead, set argument to the path for the executable.
# tmpDir: specify a temporary directory to be used. default: [$TMPDIR]
#
# outPath: specify the name of the results folder.
# In case you refer to prior modules (with fromGaia, fromRhea, etc.)
# these results must be in the same folder as well. default: results
#You can choose any of the following paths to be executed:
PCA
To perform PCA, you need to have PCAngsd pre-installed. To properly install, activate the conda environment and run:
git clone https://github.com/Rosemeis/pcangsd.git
cd pcangsd
pip3 install -e .This will make sure, PCAngsd can be called with the command pcangsd system-wide.
If you perform PCA, and no MajorMinor file exists (either in default location or defined with maMiOut), the ATLAS-Pipeline will automatically produce GLF and Major Minor files.